研究表明,约有60%的听损与遗传有关,目前已发现的听损相关基因近300个。在正常人群中约有5%-6%的人至少携带一种听损基因。
你可能会问:
“哪几个听损基因是最常见的?”
“我会不会携带听损基因?”
“遗传的概率有多大?”
全国性聋病分子流行病学调查结果显示:
21.01%的聋人携带GJB2基因突变,是中国最常见的致聋责任基因;大前庭导水管综合征的责任基因SLC26A4是中国第二高发的致聋基因,其突变检出率达12.7%;4.51%聋人携带线粒体12S rRNA基因突变,为药物性听损群体的主体。
接下来,让我们一一了解中国人常见的三大听损基因。

GJB2基因—最常见的先天性致聋基因
GJB2基因突变在1997年首次被发现,它在儿童语前聋中占20%,在儿童非综合征听损(NSHI)中占40%,是中国人最常见的致聋基因,在亚洲人群中最常见的突变位点为235delC。
致聋原因:
研究发现,GJB2基因编码一种称为“间隙连接的β2蛋白”,这种连接蛋白通过调控钾离子水平来维持相邻细胞之间的信号传导分子的转运。
GJB2基因在人类耳蜗中高度表达,形成听觉的过程中需要将声波转换为神经冲动,这种转化涉及许多过程,包括维持内耳中适当水平的钾离子,突变的GJB2基因会对耳蜗的正常的听觉功能造成严重影响。
听力损失表现:
GJB2相关性听损一般为先天性,双耳同时受累,听损程度呈对称性,少数表现为不对称性,也有单耳受累报道。GJB2基因突变造成的听力损失程度从轻度到集中度不等,大多表现为重度或极重度听损。
遗传方式:
GJB2基因突变属于常染色体隐性遗传,这也就意味着两个携带该突变基因的正常父母有概率会生出一个患儿。
孩子听损还是不听损由两个等位基因决定(A和a),两个等位基因一个来自父亲,一个来自母亲,生育的时候父母各自随机出一个等位基因“拼成”孩子的一对等位基因。
举个白话浅显的例子:
A是显性的,a是隐性的,也就是说A是“大哥”,a是“小弟”。
有A在的时候,a听A的;没有A的时候,“小弟”a可能就“胡作非为”了。
在这里,GJB2基因突变属于常染色体隐性遗传,“大哥”A是听力正常的,“小弟”a是听力不正常的。
当“大哥”A不在,而两个“小弟”a聚在一起的时候,它们就合伙捣乱,把孩子的耳朵“关上了”。
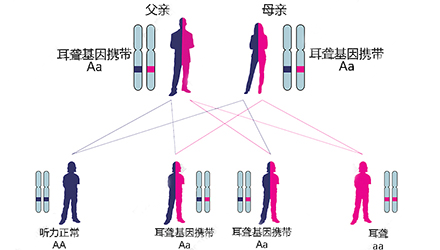
假设其他听损相关基因均正常,不考虑概率极小的自身突变,仅讨论GJB2基因:
父母中只要有一个不携带该突变基因(AA+AA/Aa/aa)——孩子100%正常;
父母均携带该突变基因(Aa+Aa)——孩子75%正常;
父母中一个携带该突变基因而一个听损(Aa+aa)——孩子50%正常;
父母均听损(aa+aa)——孩子0%正常。
怎么办:
由于GJB2基因突变造成的听力损失程度多为重度或极重度,并且听觉系统的结构基本正常,常提示人工耳蜗植入效果良好。
最好的办法是通过新生儿听力筛查及听损基因筛查尽早发现并确诊病因,尽早进行人工耳蜗植入,以保证听觉言语能力的正常发展。
SLC26A4基因--大前庭导水管相关致聋基因
SLC26A4基因定位于人类染色体7q31,SLC26A4基因和大前庭导水管综合征相关突变位点的发现,证实SLC26A4是大前庭导水管综合征的责任基因。
我们平时提到的“一巴掌打聋”、“一跤摔聋”其实都与SLC26A4基因突变有关,绝大多数大前庭导水管综合征都是SLC26A4基因突变惹的祸。
致聋原因:
SLC26A4基因编码一种叫“Pendrin”的跨膜转运蛋白,在机体离子成分平衡的维持中发挥重要作用。
在内耳,Pendrin表达于内淋巴管、内淋巴囊、椭圆囊、球囊等处,异变的蛋白将对这些结构的正常生理功能产生影响,引发听损。
听力损失表现:
SLC26A4基因突变导致的大前庭导水管综合征的典型表现为儿童时期的听力损失,90%的患者为双侧性,听力损失程度不一,可表现为接近正常或重-极重度。
病程可为稳定性、进行性或波动性,听力可逐步下降至全聋;跌倒、撞击等行为或无外界影响都可能引发听力的下降。
遗传方式:
SLC24A6基因突变属于常染色体隐性遗传,其遗传方式同上面的GJB2基因突变。
怎么办:
通过基因筛查可以在轻-中度听损中筛选出听力较好的患儿,对其正确指导,避免跌倒、撞击等外界因素,有望保存参与听力,使患者适应正常教育环境和社会。
有残余听力的病人可佩戴助听器,听力损失达到重度或极重度应考虑植入人工耳蜗。
线粒体12SrRNA基因—药物性聋相关致聋基因
线粒体12SrRNA基因是药物性聋直接相关的责任基因,约有20%-30%的药物性聋与其相关,中国人最常见的突变位点包括A1555G、C1494T等。
可以说12SrRNA基因突变是“一针致聋”的罪魁祸首。
致聋原因:
12SrRNA基因的突变会使得在蛋白质合成的过程中形成与氨基糖苷类药物结合的位点,从而影响线粒体蛋白质的合成而导致药物性聋。
也有研究表明在一些未使用听损药物的听损患者中,突变的12SrRNA还可能会使线粒体发生应激反应,启动内耳细胞的凋亡程序,从而导致听损。
听力损失表现:
听力损失通常发生在使用过氨基糖苷类药物后的3天到3个月左右,临床表现通常为双侧、重度或极重度且不可逆的高频听力损失。
也有部分该突变基因携带者在没有服用氨基糖苷类药物的情况下出现听力损失。
遗传方式:
线粒体12SrRNA基因属于母系遗传。由于在受精卵时期,线粒体都来源于卵细胞,这也就意味着该遗传和父亲无关(无论父亲是否携带该突变基因),只要母亲是携带该突变基因的患者,就会遗传给后代。
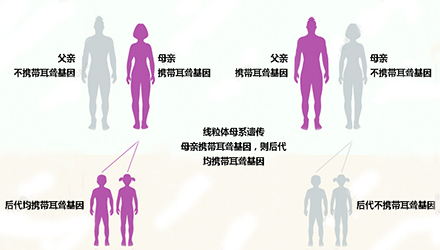
母亲正常——孩子100%正常
母亲携带该突变基因——孩子100%携带该突变基因
怎么办:
通过听损基因筛查可以早期发现药物性聋高危人群,终生避免使用氨基糖苷类药物及其他耳毒性药物,防止听损的悲剧发生。
如用药后发现听力下降,应及时到医院治疗。

听损基因检测—一次检测,家族成员终生听力健康受益
相信你已经对中国人的三大常见致聋基因有了基本了解。 听损基因的来龙去脉让我们更加清楚听损基因检测的必要性和重要性。
还是如前几期“听损基因”文章所述:“防范于未然”好过“亡羊补牢”,早检测,早发现,早干预,别让听损代代传!
对于听损患者及有听损家族遗传史的患者来说,常见听损基因检测对国人常见的几大听损基因的高发突变位点进行全面检测,明确听损的原因,从而有效避免药物、头部碰撞等因素致聋,以及科学遗传咨询,避免或减少下一代再出现遗传性听损。
近年来,基因检测技术发展颇为迅速,样本取材(即DNA取样)已不仅限于血液,口腔粘膜拭子取样简单、无创,已被认定为可靠的基因检测采样方式。
这意味着,用一个类似于“棉签”的拭子,在口腔里刮拭就可以采取DNA做听损基因检测了。